Slurm作业提交
Slurm作业通常分为交互式和批量式两种。交互式作业通常用于代码编译、脚本调试、交互式计算等工作。长期后台计算的任务通常以作业脚本的方式进行批量提交。
交互作业
集群的登录节点设置有资源限制,请勿在登录节点进行大量计算。
集群的计算节点默认不允许用户直接登录,对需要交互式处理的程序,在登录到集群后,使用salloc命令分配节点,然后再ssh到分配的节点上进行处理:
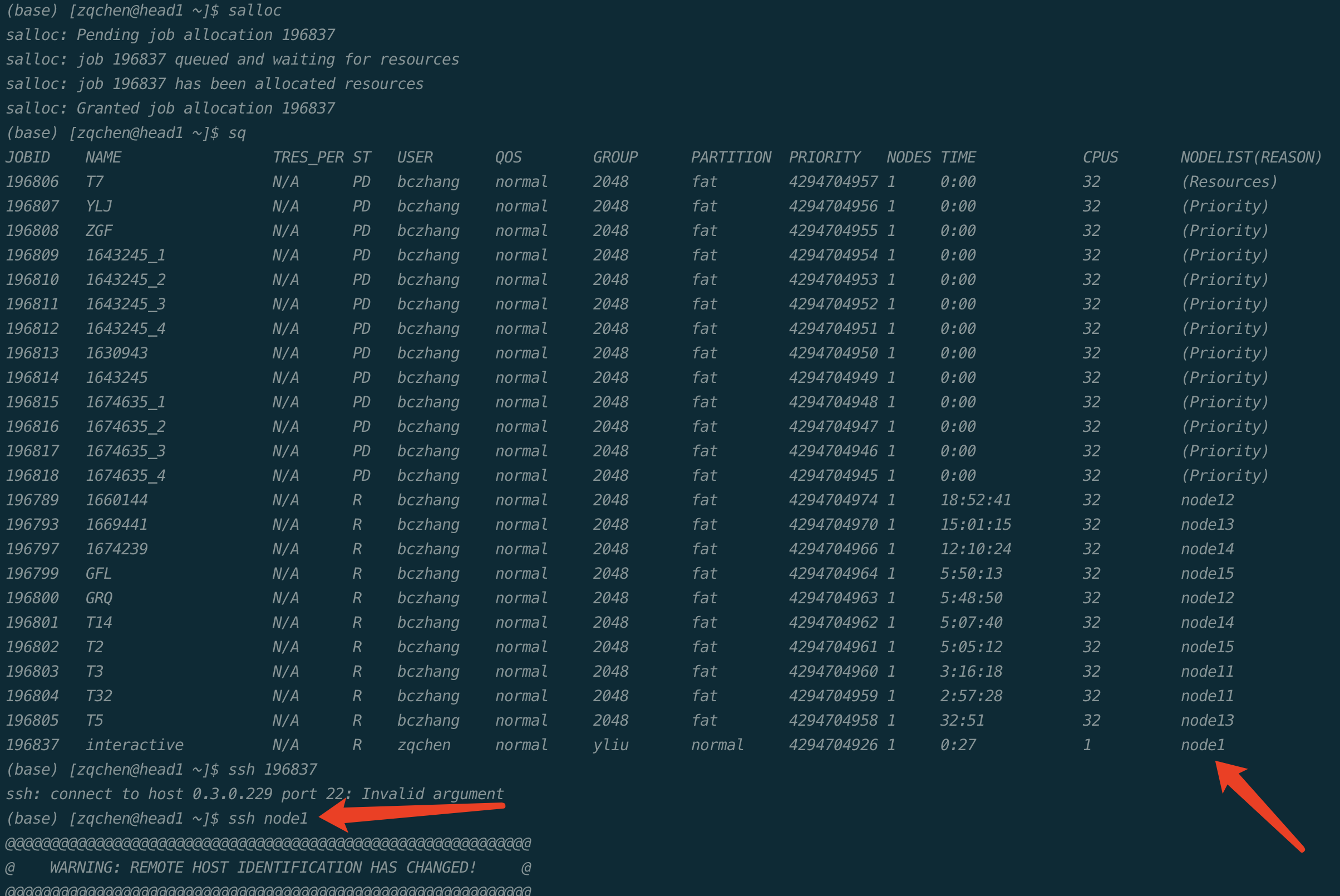
这里注意一下,我们可以直接ssh到我们的节点,区别于上医的服务器,我们可以直接进行ssh。如:
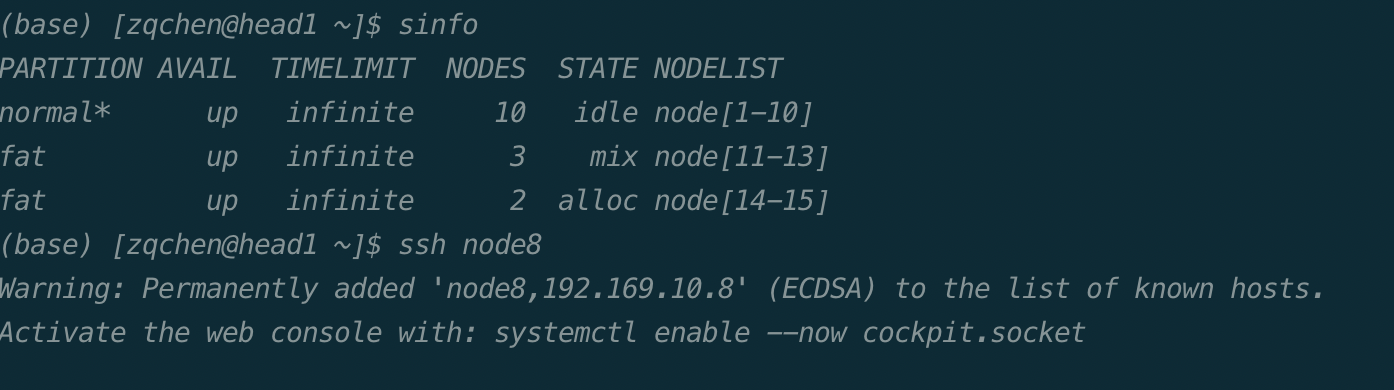
计算完成后,使用exit命令推出节点。
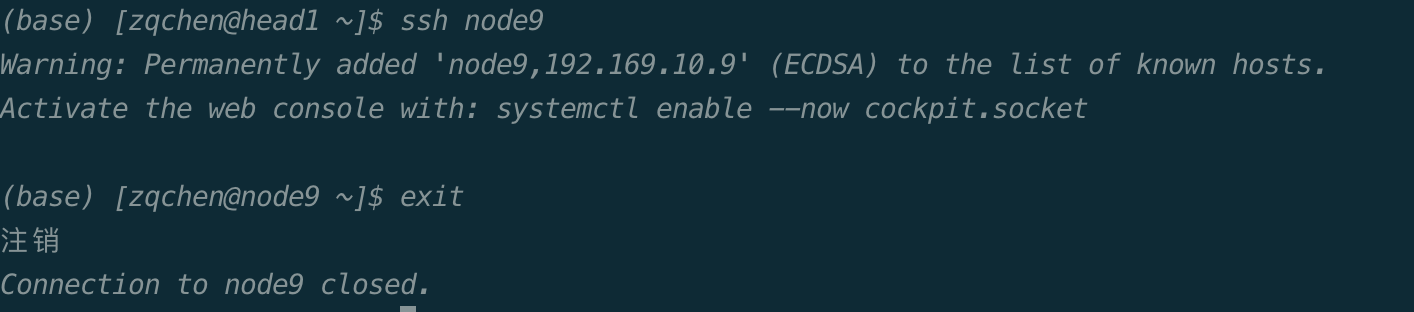
exit两次,第一次exit是从计算节点退出到登录节点,第二次exit是释放所申请的资源。
批量作业
可以通过将程序执行命令放入作业提交脚本,并通过sbatch命令作业提交的方式在集群中进行计算。
一个简单的脚本示例如下:
1 2 3 4 5 6 7 8 9 10 |
#!/bin/bash
#SBATCH --job-name=JOBNAME
#SBATCH --nodes=2
#SBATCH --ntasks-per-node=10
#SBATCH --time=2:00:00
### 程序的执行命令
python count.py
|
注意:
上述中JOBNAME:作业的作业名。
上述中nodes指定该作业需要2个节点数。
上述中ntasks-per-node每个节点所运行的进程数。
上述中time作业最大的运行时间。
第一行表示这是一个bash脚本,第3-6行以#SBATCH开头的命令表示这些是需要slurm系统处理的参数。
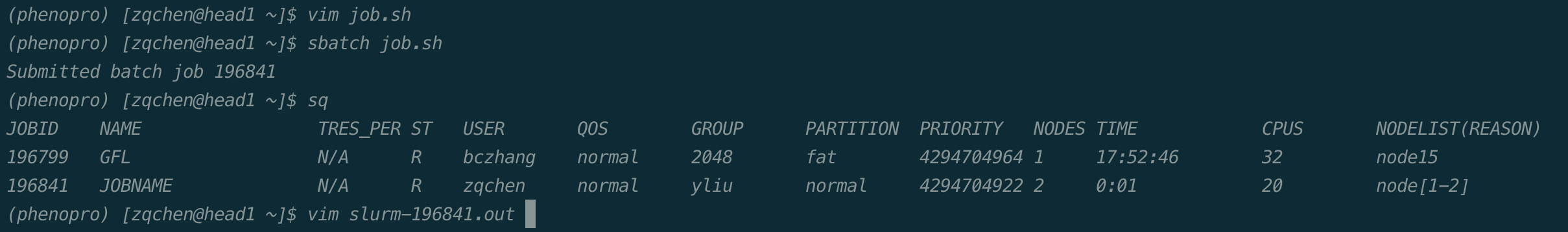
如下图所示,通过sbatch+作业脚本名提交作业,系统会返回作业编号,通过squeue命令可以看到作业运行状态,等作业执行完成后,默认会把程序的输出放到slurm-作业编号.out的文件中,可通过该文件查看程序的输出。
GPU集群作业提交
如果是GPU集群,需要在作业脚本中增加--gres=gpu:<number of card>参数。例如#SBATCH --gres=gpu:2,意味着指定2张GPU卡数。
以下为GPU作业的一个示例:
1 2 3 4 5 6 7 |
#!/bin/bash
#SBATCH --job-name=gpu-example
#SBATCH --nodes=1
#SBATCH --ntasks=16
#SBATCH --gres=gpu:1
### 程序的执行命令
python test.py
|
注意:
上述中job-name:作业的作业名。
上述中nodes:该作业需要的节点数。
上述中ntasks:该作业需要的CPU数。
上述中gres:该作业需要的GPU数。
GPU集群中提交作业时,需要在srun 或 sbatch命令中增加参数-s,或者 --oversubscribe。表示允许与其它作业共享资源。
例如:
1 | $sbatch -s job.sh
|
常见提交作业参数参考
参数 |
说明 |
|
设定作业名称 |
|
设定作业需要的节点数。如果没有指定,默认分配足够的节点来满足 |
|
设定每个节点上的任务数。要和``–nodes=<n>``同时配合使用。 |
|
设定最多启动的任务数。 |
|
设定每个任务所需要的CPU核数。如果没有指定,默认为每个任务分配一个CPU核。一般运行OpenMP等多线程程序时需要,普通MPI程序不需要。 |